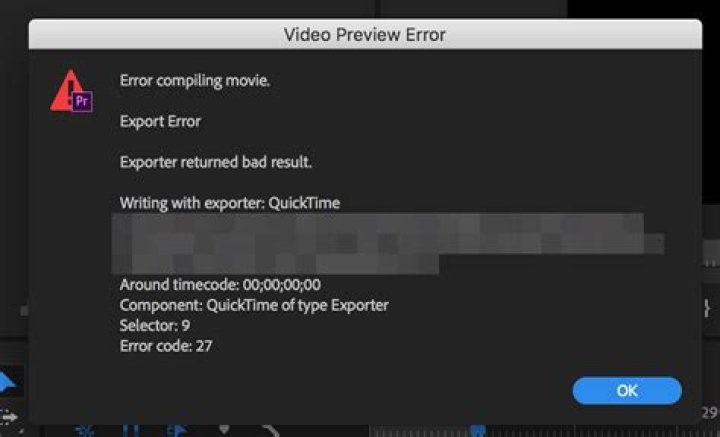
I am new to R and I am trying to make an arrowplot. However, the basic gggenes set3 colour theme only has 12 colours and I need more.
I want to assign a group of genes with colour (eg, glycosyltransferases all red and methyltransferases all blue)
I have added an extra column to my df named "colour" and assigned each gene with one hex code (#c1ffc1) - just to test that all genes could change colour before going through and assigning the ones for glycosyltransferases etc - I managed to get it to change colour once and now it isn't working?
Here is the code example with three genes
#add colour column to assign to genes > colour <- c("#c1ffc1") > df1$colour <- colour > #change colour > library(ggplot2) > library(gggenes) > ggplot(df1, aes(xmin = start, xmax = end, y = molecule, fill = colour)) + + geom_gene_arrow() + + geom_gene_label(aes(label = gene)) + + facet_wrap(~ molecule, scales = "free", ncol = 1) + + theme(legend.position="top") + xlim(0,37841) + scale_fill_discrete(name = "gene", labels = c("VanH", "VanA", "VanX")) molecule start end strand gene orientation colour KJ364518.1 2314 3345 reverse vanH 1 #f15854 KJ364518.1 3347 4387 reverse vanA 1 #f15854 KJ364518.1 4384 4992 reverse vanX 1 #f15854 KJ364518.1 6334 7125 reverse ajrR 1 #faa43a KJ364518.1 7246 8097 reverse pdh 1 #5da5da KJ364518.1 8410 10272 reverse tri 1 #b276b2 Thanks so much in advance, Lucy
62 Answers
Your code appears to work fine, the only thing is that you're providing literal colours to the mapping instead of fill = your_classification_here. To show literal colours you can use scale_fill_identity().
library(ggplot2) library(gggenes) txt <- " molecule start end strand gene orientation colour KJ364518.1 2314 3345 reverse vanH 1 #f15854 KJ364518.1 3347 4387 reverse vanA 1 #f15854 KJ364518.1 4384 4992 reverse vanX 1 #f15854 KJ364518.1 6334 7125 reverse ajrR 1 #faa43a KJ364518.1 7246 8097 reverse pdh 1 #5da5da KJ364518.1 8410 10272 reverse tri 1 #b276b2" df1 <- data.table::fread(text = txt) ggplot(df1, aes(xmin = start, xmax = end, y = molecule, fill = colour)) + geom_gene_arrow() + geom_gene_label(aes(label = gene)) + facet_wrap(~ molecule, scales = "free", ncol = 1) + scale_fill_identity() 
Created on 2022-05-11 by the reprex package (v2.0.1)
4When you create your dataframe one of the columns named orientation contains the info Orientation, expressed as 1 or -1. If you add the command
forward = orientation
in your ggplot aesthetics it will apply the orientation of each gene.